Hiding in plain sight
Gaya Amarasinghe
Washington University in St. Louis
Published July 27, 2018
On a recent visit to the laboratory where he worked as an undergraduate, Gaya Amarasinghe thought about his scientific journey from student to professor. At first glance, his research interests seem to have drastically changed. Back then, he was studying signaling in cancer biology, and how oncogenes communicate within cells. Now, his group at Washington University in St. Louis investigates how some of the world’s deadliest viruses, such as Ebola, outcompete the host and cause disease.
Yet, he decided, not much has changed after all. “In the last 20 years, I haven’t done anything differently,” joked Amarasinghe, who trained as a structural biologist and biochemist. “My group now studies proteins from viruses and bacteria, but they interact with and sometimes hijack the same immune signaling cascades I studied before. I’m asking the same questions.”
Amarasinghe’s group examines how the host immune system recognizes foreign pathogens and how normal immune signaling changes in the presence of microbes.
“We ask simple questions, and we answer them using simple experiments,” Amarasinghe says. “What are the initial components in the system, what does the structure look like, and what are the dynamics of these molecules? Then we ask what happens to those properties when they interact with a virus. What does that interaction look like? How does it move? What changes?”
It was a question that first made them curious about Ebola and related Marburg viruses: How do they evade the pathogen detection system that the immune system uses to distinguish self from non-self?
Most people know these filoviruses as causes of severe, often fatal, outbreaks of hemorrhagic illness, usually in Africa, where people first catch the contagious diseases from animals.
Few viruses are as dependent upon first interactions with the host as Ebola and its cousins. Unlike other viruses with multiple ways of manipulating an immune response during an infection, Ebola and Marburg viruses begin with a knock-out punch to the immune system defenses. It happens at the earliest stage, Amarasinghe says.
The critical moment comes when the innate immune cells that function as sentinel cells in the host, such as dendritic cells and macrophages, recognize the viruses as unwanted foreign invaders—or not, as is often the case.
A key player is a crucial viral protein called VP35. Without a working VP35, the viruses cannot defend early immune responses during infection. It also blocks many steps of the host’s innate immune response, including one that ramps up the antiviral immune fight.
The first attempts to answer the question about initial recognition of the virus were anything but simple. “The hardest part was getting the material we needed,” Amarasinghe says. It was a brute force effort to purify the protein. He estimates it took the equivalent of 10-20 people-years of work in many groups, including his own, to finally isolate well-behaved VP35 by genetic recombination techniques.
In 2009, his group published the first of a half-dozen papers, which include at least 10 different VP35 structures of what the interferon inhibitory domain (or IID) from several Ebola strains and Marburg, which have different disease courses.
“VP35 IID was nothing like anyone predicted,” Amarasinghe says. “This protein is hiding the non-self signal that would otherwise get recognized by the immune system. It’s hiding the signal in plain sight.” The protein doesn’t even need to move much. It has a fold that fits neatly over the non-self signal, an end of the detection RNA found in every cell.
The group also found a fundamental difference between Ebola and Marburg viruses in how they recognize the host RNA and hide it. “They are equally virulent and pathogenic, but there is a key difference at an early stage,” he says. “Why does that matter? What happens in early stages determines the outcome of infections.” For example, Reston Ebola virus, a close cousin of the more virulent filoviruses also uses VP35 to bind RNA, but differences in other parts of the virus replication cycle make Reston non-pathogenic in humans.
Once they knew what the protein looked like in the related hemorrhagic viruses, Amarasinghe’s team helped design mutated versions to model the disease and better define the biology. The mutated viruses were immunogenic.
“Very cool, because now we have virulent and avirulent near-isogenic viruses to study the disease in animal models” he says. This knowledge can be used to develop actual vaccine candidates with different molecular backbones or using smaller subunits for safety, he says.
Most of the lab’s work happens in test tubes with non-infectious viral proteins. “We have collaborators working in biosafety level 4 that put some of these crazy ideas to work,” he says. Amarasinghe and his collaborators are testing the biological mechanisms in animal models.
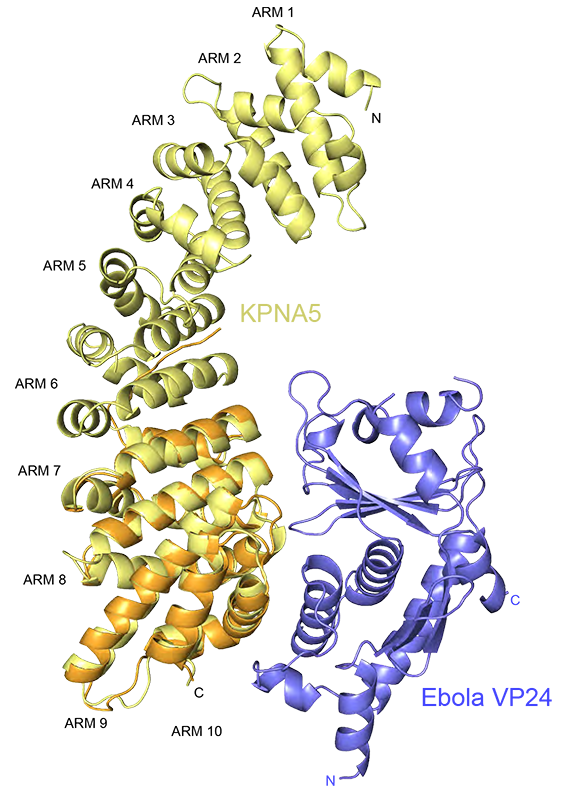
A second set of discoveries from their work helps explain why Ebola and Marburg are not responsive to interferon, a potential treatment given for many viral infections, but as it turns out it does not work well in filoviral infections. VP24, another protein encoded by the Ebola virus binds to a key cargo transporter called karyopherin, which is responsible for moving important signals, or transcription factors, into the nucleus to trigger an immune response. Viruses are known to block dendritic cells from maturing, preventing the normal interferon signaling necessary for an adaptive immune response. Amarasinghe and his colleagues have shown that VP24 also inhibits molecular signaling from therapeutically delivered interferon.
“Typically we think of a host-microbe arms race, with the host evolving away from the pathogen,” Amarasinghe says. “In this case, the place where the viral protein binds is unable to evolve away, because the host would become non responsive to its own immune signal. The virus identified an area where mutation change rate is low.”Most of the lab projects fall into one of two categories—how host factors recognize the signature or foreign pathogens, and how host signaling changes in the presence of a microbe.
The host stress response to a virus, and not the virus itself, could be responsible for some diseases and point the way to better treatments. This could be the case with the human respiratory syncytial virus (hRSV), an infection most kids acquire by age 2. It can be fatal to infants and the elderly. It is in the same family as another emerging infectious disease people catch from animals is Nipah virus, an often fatal infection that causes encephalitis and severe respiratory distress.
Amarasinghe and his colleagues purified and solved the first crystal structure of an hRSV protein known as NS1. They reported their findings in 2017 in Nature Microbiology. They found a duplicated structural fold that suggests NS1 may have a greater role in regulating host responses than previously appreciated. “It’s not quite falling off log, but it’s close,” he says about the functional revelations.
In the future, Amarasinghe says his lab will continue to explore what drives infection in the early stages from host response point of view, aiming to understand factors that determine outcome in longer term persistent infections. The knowledge may be useful in other bacterial and viral systems, including the effect of co-infections on outcomes. “None of these exist in isolation,” he says.
Amarasinghe starts many days as early as 4:00 am and most mornings with a long run or a bike ride. It keeps him in shape for occasional long-distance mountain biking or skiing.
By his name alone, Amarasinghe may have been destined to work with proteins and RNA. Every letter in his full name represents an amino acid. He pointed out that his first name, GAYA, also forms a tetraloop (guanine-adenine-pYrimidine-adenine).
-Carol Cruzan Morton