Data in Motion
Nozomi Ando
Cornell University
Published September 29, 2023
For nearly a century, protein crystallography has focused on certain X-ray signals to reveal the atomic architecture of proteins. But even a structure solved to the reigning gold standard of X-ray diffraction does not always disclose the secrets of a protein’s actions.
To uncover the dynamic details, Nozomi Ando has turned to an additional set of blurry signals usually tossed aside in the early stage of processing data gathered at the end of a beamline.
“Instead of using the signal that's used for crystallography, we’re using everything else that people have assumed is junk,” says Ando, who runs a laboratory at Cornell University in New York focused on the structure and dynamics of enzymes. “The junk signal is actually the signal that tells us how these fluctuations are happening on a very subtle basis inside the protein.”
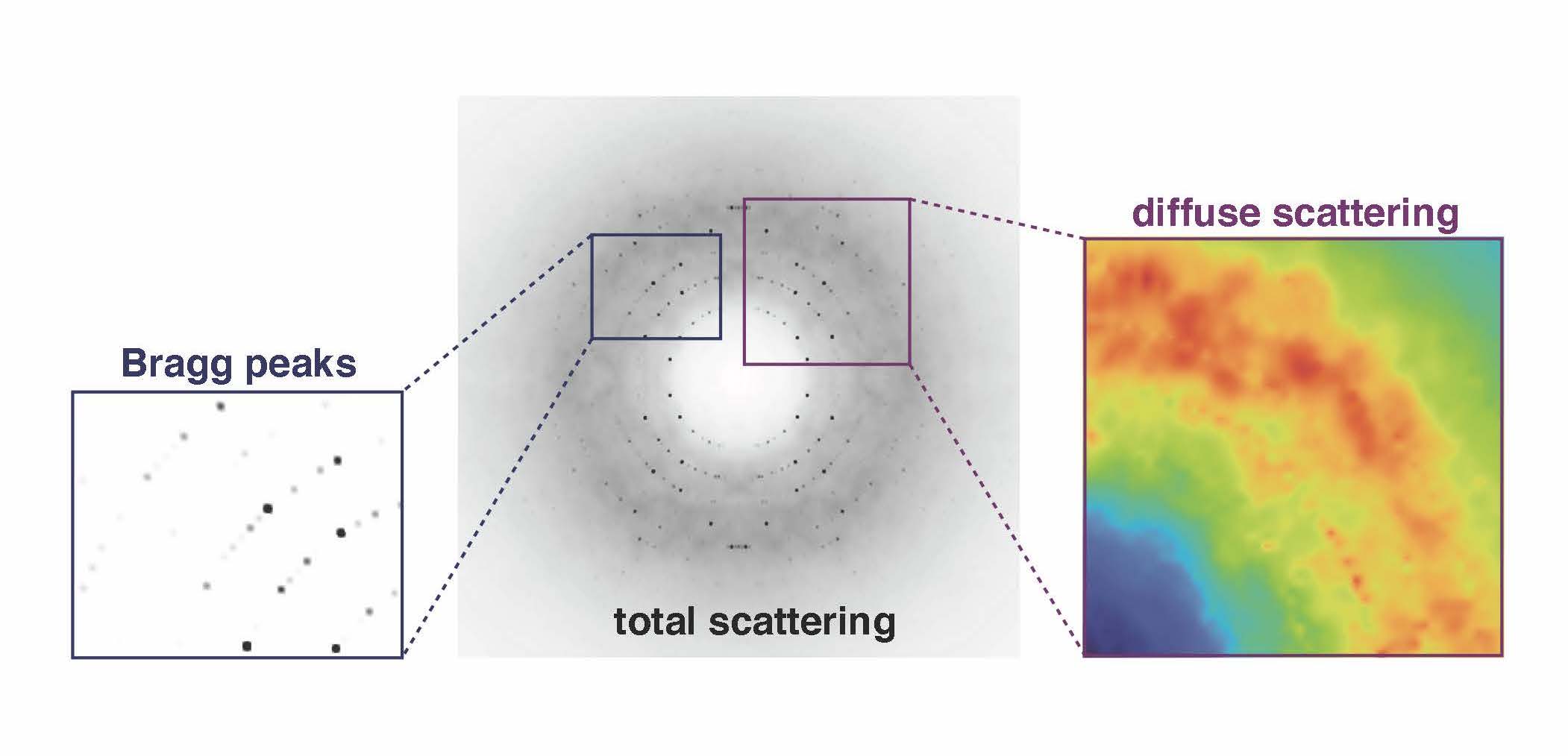
The junk signal, called diffuse scattering, usually is edited out in favor of the more authoritative Bragg intensities that ping off the lattice of ordered proteins in a crystal. The Ando lab is developing tools and techniques to probe those fuzzy weak signals for information about how the proteins move.
It’s not completely obvious that crystallography can generate dynamic data. After all, the exquisitely detailed models arising from Bragg signals give the impression of a static molecule. But in reality the atomic positions of the model are averages of proteins that can’t stop fidgeting.
“You just can't stop a molecule from moving unless you're at zero Kelvin,” Ando says. “It's always moving even inside of a crystal lattice.”
The idea that the diffuse signal has potential to animate crystal structures goes back at least three decades, but those signals have been difficult to measure and interpret due to major technical and theoretical limitations.
“The frontier of structural biology is definitely headed in the direction of trying to understand a protein more as a movie as opposed to its static nature,” she says. “But it's still really hard for structural techniques to capture molecules that can’t stay still.”
Ando launched her independent career just as the tools to capture the signal—direct detectors—were becoming common. And her training in X-ray scattering had given her experience with disordered samples and messy datasets.
With its first postdoctoral fellow, Steve Meisburger, and a visiting faculty member, David Case, the Ando lab embarked on a complicated mission to create a diffuse scattering map for triclinic lysozyme, an enzyme that breaks down bacterial cell walls.
“Suddenly, we were seeing details in the diffuse scattering signal that had been hypothesized but not ever clearly seen before,” Ando says. The team found two main types of signals that need to be delineated in diffuse scattering analysis (Nature Communications, 2020). The brightest signal came from protein-protein interactions in a lattice that itself moves. Then there was a much weaker signal coming from motions within the protein.
“That was really exciting for us because, for the first time, we were able to not only get an accurate picture of the signal, but once we saw it, we could explain it in terms of very simple physics with very good statistics,” she says.
More recently, they developed a robust workflow with validation that others can use to isolate the internal motion signal from total scattering data (Nature Communications, 2023).
“The key takeaway is that any atomic motions implied by refinement of a structure should agree with diffuse scattering,” she says.
Ando was born in Denver, Colorado, to Japanese parents who had moved there for her father’s graduate studies in materials science. The family moved to the Boston area for her father’s postdoctoral work at the Massachusetts Institute of Technology (MIT). In preparation for a return to Japan, where the school year starts in Spring, they enrolled Ando in first grade early so she wouldn’t fall behind and began tutoring her in the universal language of math.
“That was actually my gateway into science,” she says with hindsight. “Making sure that I didn't get behind in school meant that I was always ahead in math.”
At the time, if she couldn’t play outside, Ando was more interested in painting and drawing. After nearly three years in Japan, the family decided Ando and her younger sister would have more opportunities in the United States. They returned and settled in the Boston area. In high school, many of Ando’s friends’ parents were professional musicians, and her artistic interests turned to singing.
She applied to the one college she knew from her dad’s postdoctoral days, MIT. At the time, she assumed biology was all about memorization, so she chose to be a physics major. Within physics, she tried to find her place. It became quickly apparent that there were very few women in physics—11 percent of undergraduate physics majors at the time. In her science classes, she had one female professor in four years.

Then she found where she belonged—a lab, headed by Toyoichi Tanaka, who was using polymer hydrogels as a model system for proteins. The gels underwent enormous phase transitions, enabling them to swell or contract by up to 1,000 times in volume.
“There were just so many different flavors of physics I was exposed to, and I just couldn't get excited about all the other ones, somehow, and then biophysics just suddenly spoke to me,” she says. “And I've been in love with proteins ever since.”
Sadly, at the end of Ando’s junior year, Tanaka died unexpectedly in his 50s. Ando stayed in the lab doing research without an advisor. She wrote a small grant to the department to buy equipment for her experiments. She sang with the MIT choir and sang solos with the MIT orchestra.
A graduate school scouting tour of the X-ray synchrotron facility at Cornell, plus a chat with director Sol Gruner, clinched the next step. A lifelong fear of X-rays suddenly fell away when Ando walked through the tunnel of the giant machine, ducking under cables and gazing at the flickering LED lights.
The Gruner lab emphasized developing new techniques and tools. There, Ando built her own small angle X-ray scattering sample cell that could be used under high pressure to study the interaction of proteins and water in model systems.
Gruner invited Ando, a lyric soprano who favors operatic arias and poetry set to music, to sing at two synchrotron site visits by the National Science Foundation. At one, Lois Pollack, now a faculty colleague at Cornell, was the page turner for Ando’s performance. (Another faculty colleague at Cornell is her husband Buz Barstow, a former Gruner labmate.)
Ultimately, Ando wanted to understand more about the proteins themselves. So for her postdoctoral work, she moved into chemistry, returning to MIT to study metalloenzymes in the lab of Catherine Drennan.
Ando brought her X-ray scattering experience to a lab full of crystallography expertise. “And together, we could answer new types of questions,” she says.
Small-angle X-ray scattering is a low-resolution technique that allows samples in solution to move around, while crystallography is a high-resolution technique that generates a clearer but more static picture of a structure, Ando says.
“And that combination is really powerful,” she says. “if you have high resolution information, as well as information about how conformational changes occur, you can start seeing the behavior of the protein. And this gives you some insight into the landscape of proteins, like all the different conformations that proteins can adopt.”
Ando started her own lab in 2014 in the chemistry department at Princeton. She combined her two backgrounds—X-ray physics and the biochemistry of enzymes—with the ultimate goal of understanding how a protein’s sequence is connected to its structure, dynamics, and function.
In 2018, the Ando lab moved to Cornell, where most of their X-ray experiments were being done. The move also allowed the research program to grow in a new direction that incorporates bioinformatics and cryo-electron microscopy.
Soon after, the Ando lab started a collaboration with Colin Jackson’s lab at the Australian National University to study the evolution of ribonucleotide reductase, an enzyme that is used by practically all organisms to make the building blocks of DNA. Using bioinformatics, X-ray scattering, and cryo-EM, they discovered a new group of ribonucleotide reductases with implications on how life adapted to oxygen on the early planet. This study was published in eLife (2023) with three graduate students from the two labs, Audrey Burnim, Matt Spence, and Darren Xu.
More recently, the Ando lab team led by graduate student Maxwell Watkins, combined X-ray scattering and cryo-EM, as well as structure prediction by AlphaFold, to analyze an enzyme constructed like four beads on a string (PNAS, 2023). The light and oxygen-sensitive enzyme had to be studied in the dark in an anaerobic chamber. They found that the floppy enzyme has a default conformation that springs into action when the right substrate binds. Remarkably a deep dive into the AlphaFold database turned up the same dynamic conformation, suggesting that it is conserved even when the protein sequence varies.
“Ultimately, we’re trying to understand how a protein works,” she says. “To me, that mechanism involves both structure and dynamics. Enzymes can change their structures or the way they move in response to the environment. And I think that's really amazing, because that's part of what makes proteins seem like they're alive. The combination of X-ray methods and cryo-EM has been extremely powerful in studying their structural dynamics. By incorporating bioinformatics, we gain another knob to turn and learn about how protein behavior changes as a function of protein sequence.”