Structures Solved Simply
It used to be that to book a trip you'd first need to call every airline to compare flights. Then you'd need to find good hotel deals. Then you'd have to revisit the flights. And so on. The same trial and error approach also used to hold true for structural biology. Frequent failures made scientists all too familiar with square one.
Now, however, what Orbitz and Expedia have done for travel, Phenix has done for structural biology.
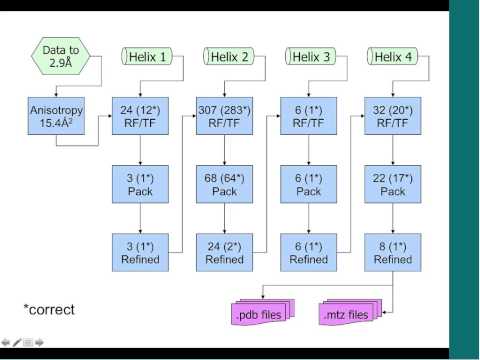
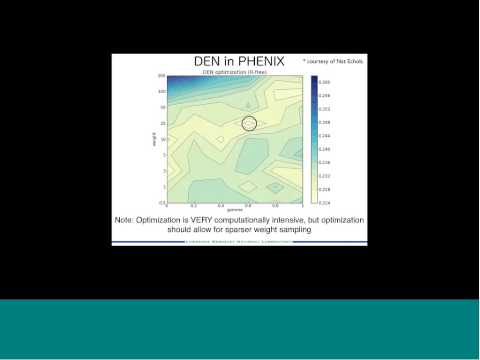
Phenix helps investigators solve Xray crystal structures using multiple approaches, including molecular replacement and experimental phasing. It makes things easier by streamlining these multi-step procedures programmatically. Experimental data goes in, and a solution comes out.
Phenix Wizards perform the magic. For instance, the autosol program drives experimental phasing …
Read the full story here.